
epitope_diversity: Estimating the mutation diversity within a genomic region (e.g. epitope)
Last modified:Introduction
When deep sequencing data is available, sometimes it is interesting to check the mutation diversity of reads within a specific genomic region. A frequent example would be studying the mutation diversity of a immune epitope region.
I first came across this problem in a study on flu virus population under T cell–activating vaccination. At that time, I wrote a R script diving into the alignment BAM file and calculated the Shannon entropy and Gini-Simipson’s index for epitope haplotypes observed in the alignment. One main problem for the R scripts is that it runs too slow and took too much memory, so I recently developed a likewise tool called epitope_diversity using Rust, and I believe this tool is more fast-running, memory efficient, and easier to use. For the installation and usage of the software, please check this link.
In the below section, I will briefly describe methods used for estimating mutation diversity.
Methods
There are two levels of methods for calculating mutation diversity:
- Nucleotide/Locus level
Useful when haplotypes are not available. For example when you only have the vcf files (mutations for discrete genomic sites), or when you want to calculate the whole-genome genomic diversity, but you only have short-read sequencing data so you don’t know the full-length haplotypes. - Haplotype level
Good to use when haplotypes are available.
Nucleotide/Locus level
Basically we calculate the diversity for every nucleotide position (locus), then take average over the whole epitope region. For details please refer to this paper.
Supposing that full haplotype information for the virus is not available, a genome-wide measure of entropy may then be calculated, computing the mean of this statistic across all sites (McCrone and Lauring 2016).
Lei Zhao, Christopher J R Illingworth, Measurements of intrahost viral diversity require an unbiased diversity metric, Virus Evolution, Volume 5, Issue 1, January 2019, vey041, https://doi.org/10.1093/ve/vey041

Haplotype level
We first tried to count the frequencies of different haplotypes, then we calculate the metrics like Shannon entropy or nucleotide diversity. Details please see this paper.
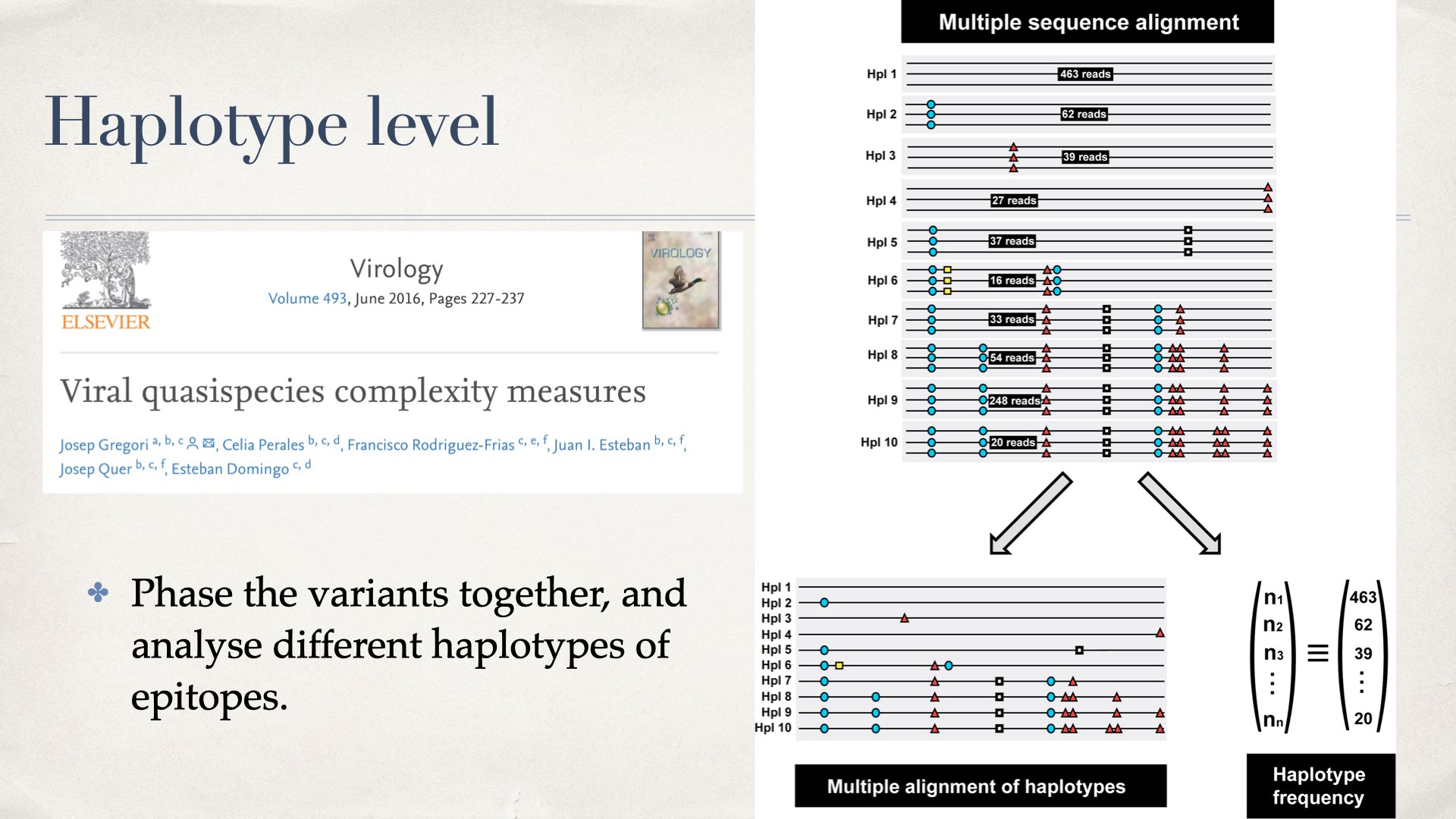
Detail steps (Haplotype level)
The below slides show the essential six steps in measuring mutation diversity using epitope_diversity:
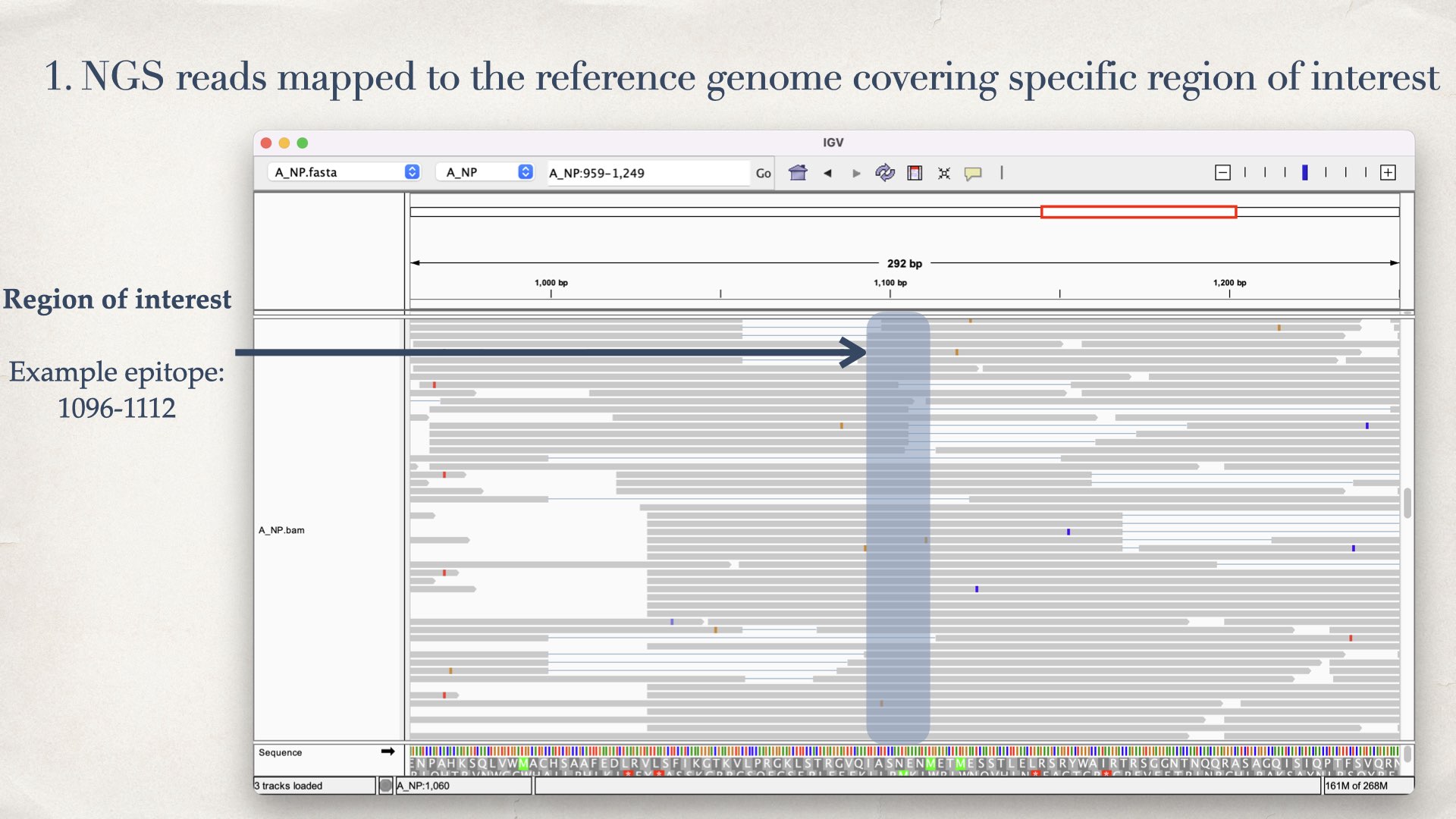
- NGS reads mapped to the reference genome covering specific region of interest.
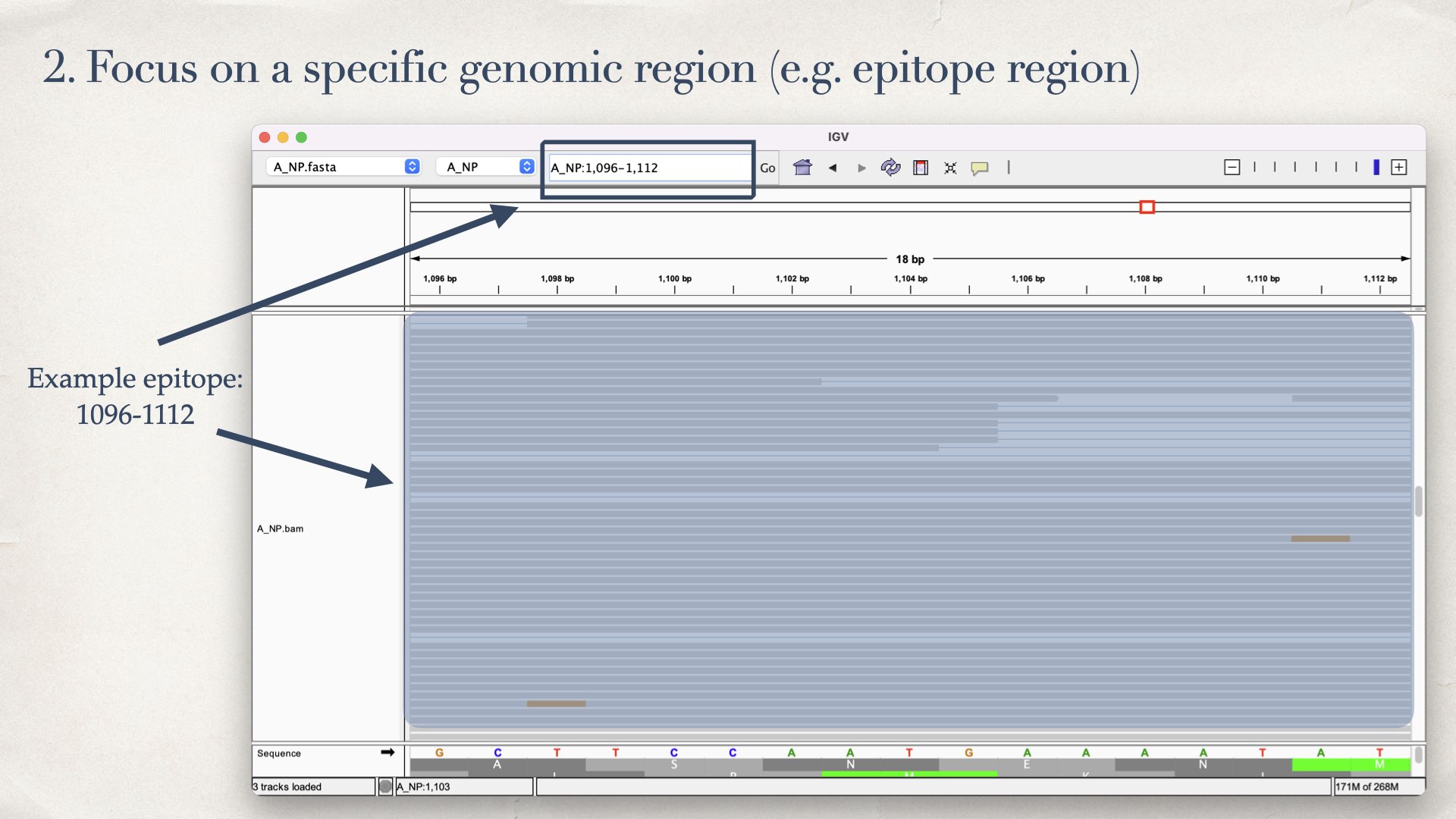
- Focus on a specific genomic region (e.g. epitope region)
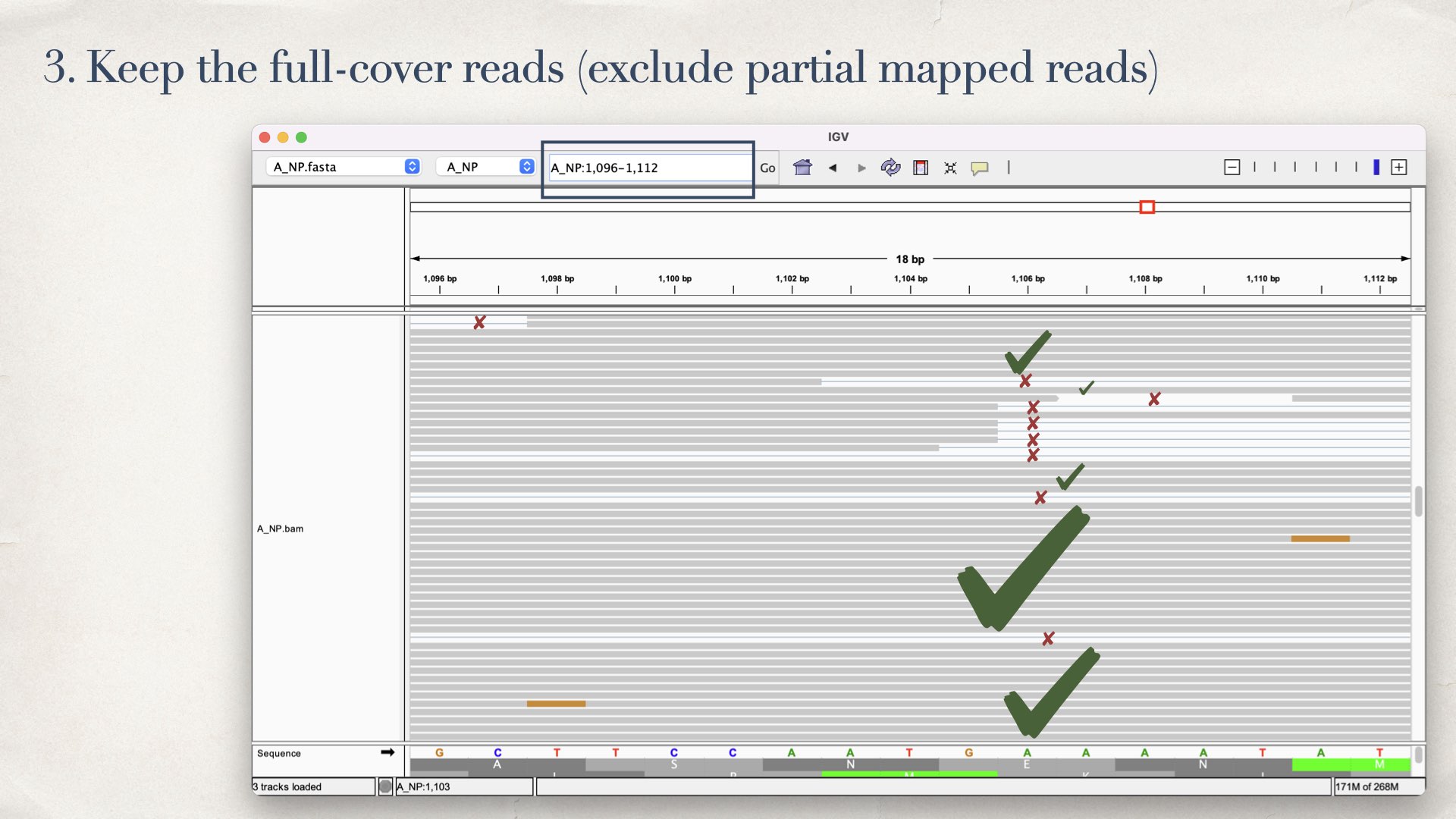
- Keep the full-cover reads (exclude partial mapped reads)
- Identifying different haplotypes from the full-cover reads (using HashMap)

- Count the number of reads for every haplotype
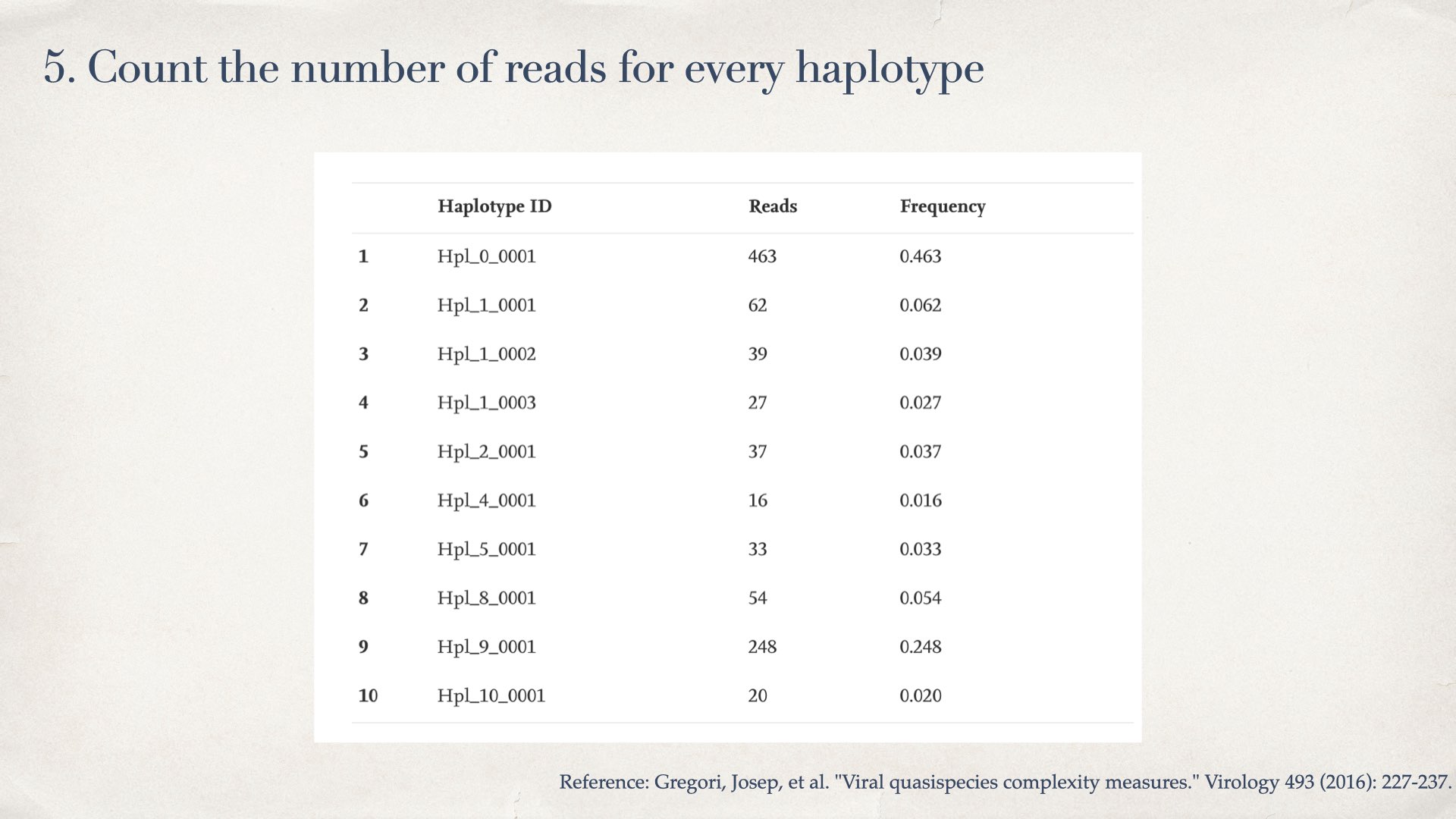
- Calculate the diversity by frequencies of haplotypes (Shannon entropy), and nucleotide difference between haplotypes (population nucleotide diversity)

Please feel free to contribute to the project and raise issues here.
Tags: Molecular evolution, Programming
Categories: My tools
Comments