
Paper digest: Selection on synonymous sites: the unwanted transcript hypothesis (Nat Rev Genet, 2024)
Published: Post views:The paper titled Selection on synonymous sites: the unwanted transcript hypothesis by Laurence D. Hurst et al. discussion current understanding why synonymous mutations are not neutral. The part related to CpG dinucleotides is particularly interesting.
The unwanted transcript hypothesis
Widespread spurious transcripts
- In the human genome, transcription factor binding sites are everywhere, nearly all the DNA of the human genome is transcribed.
- More than half of human DNA is derived from transposable elements, 83% of lncRNAs have exonized transposable elements.
- The transcription machinery is not perfect, slicing is error-prone.
- As a result, spurious/unwanted transcripts are heavily produced.
Costly spurious transcripts
- Translation are energetically demanding, and the translation machinery is limited.
- Expression of some unwanted transcripts are directly toxic, some can interfere with the expression of other genes.
The solution
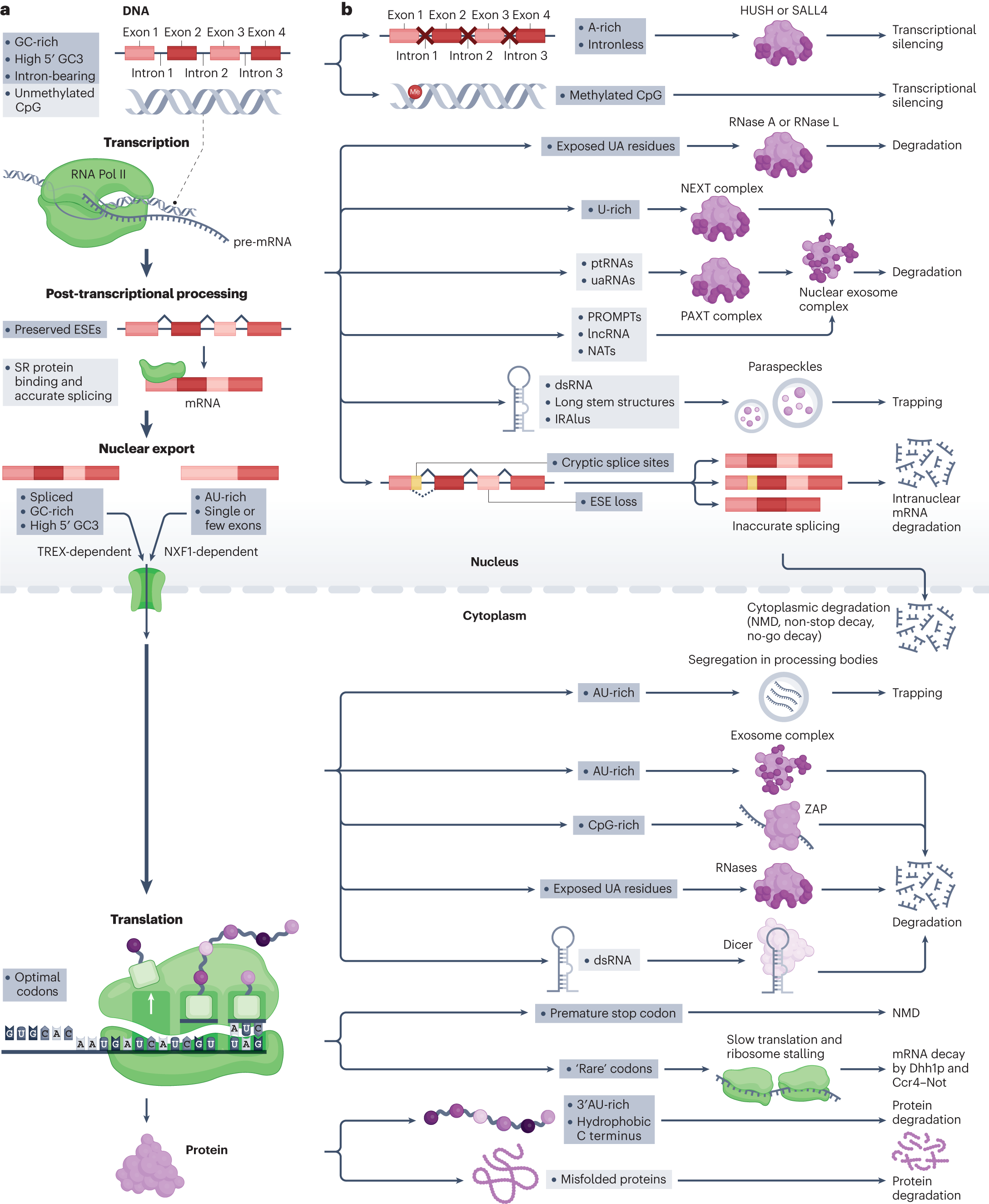
- First to curtail their creation, e.g., gene methylation in humans to reduce rates of spurious intragene transcriptional initiation from cryptic promoters.
- Second, to have QC mechanisms to degrade (e.g., by ribonucleases (RNases)) or physical isolation (e.g., preventing nuclear export) unwanted transcripts.
- Details in Figure 1.
{kind=link}
Selection on codon usage
- CpG -> metCpG -> metTpG: methylated CpG is hypermutable (10-fold higher mutation rates).
- GC -> AT mutation bias: Even in the absence of methylation, GC→AT transitions are more often than the reverse. Non-coding regions, which aren’t under strong selective pressure, will accumulate more A/T bases.
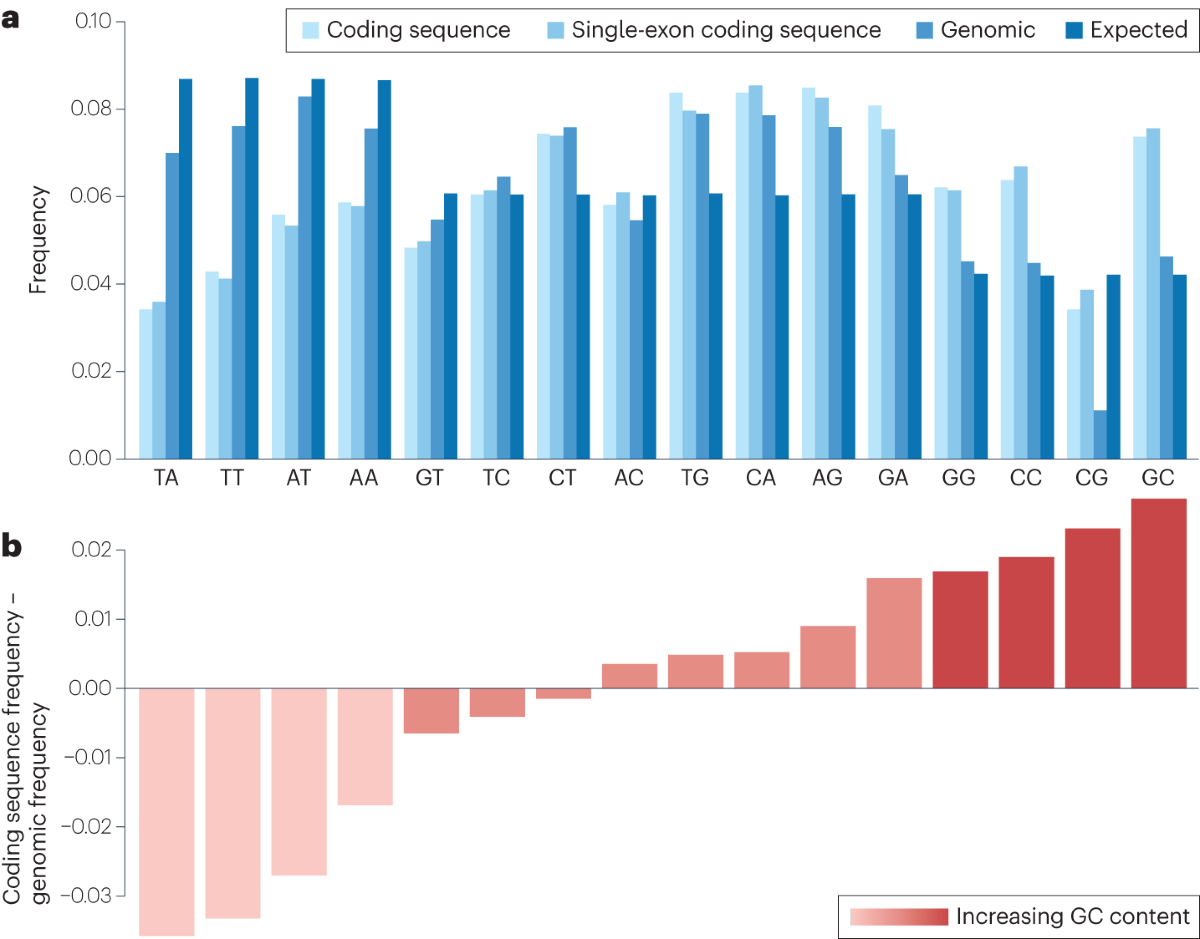
- Check the Figure 2 for the dinucleotide frequencies in the human genome.
- Because of the above two reasons, the genomic sequences are generally AT-rich and CG-poor.
- However, the coding region sequences have higher GC content.
- Some of these amino acids are metabolically cheaper to produce, or avoid undesirable stops. As a result, many functional genes maintain high GC content, particularly at synonymous codon positions (e.g., GC3).
- GC-Biased Gene Conversion (gBGC) can elevate GC content in gene-rich regions by favoring GC over AT during DNA recombination. This effect can preserve or amplify high-GC “signals” in coding regions, distinguishing them from the AT-rich parts of the genome.
- Selection on synonymous sites: GC-rich is preferred in coding regions, and AT-rich sequences as recognized suspicious.
- CpG is a notable exception to the GC-rich rule. CpG are hypermutable, so in coding regions, it is bad. Cells tend to prefer GC‐rich, CpG‐poor transcripts, especially those with multiple small exons—features that mark them as native. Viruses with high-GC content often violate this rule. Transcripts that deviate from this pattern, such as **AT‐rich, single‐exon, or high‐CpG **RNAs, are more likely to be considered non‐native and subject to suppression or degradation.
{kind=link}
What can the hypothesis explain?
- High‐GC Content Promotes Expression
- Genes enriched in GC, especially at synonymous sites (like the third codon position), are recognized as native and pass quality control checks, enhancing their transcription and nuclear export.
- This explains why intronless but GC‐rich genes (including many retrogenes) achieve robust expression and why GC‐rich codons can boost protein output in transgene studies.
- Avoidance of High‐AT and High‐CpG
- AT‐rich or CpG‐rich transcripts are flagged as suspicious.
- Mechanisms like CpG methylation and the HUSH complex selectively silence or degrade such transcripts, effectively minimizing unwanted RNAs (from viruses or transposable elements) and preventing large‐exon, single‐exon, or A‐rich mRNAs from being expressed unless they have specific adaptive features.
- Splicing Fidelity at Synonymous Sites
- Humans show strong selection to preserve exonic splice enhancers (ESEs) at synonymous positions near exon boundaries to ensure accurate splicing, preventing the creation of aberrant splice variants.
- Conversely, synonymous mutations that create cryptic splice sites or disrupt known splicing signals are strongly disfavored.
- Quality Control Through Multiple Filters
- Steps such as nuclear export (handled by the NXF1 or TREX complexes), cytoplasmic processing bodies, and stress granules filter RNAs based on features like GC content, exon length, or A‐rich sequences.
- Additional checks (e.g., nonsense‐mediated decay, codon‐optimality–mediated decay) can degrade suspicious or faulty transcripts even if they reach the cytoplasm and engage ribosomes.
- Broader Parallels with Viral Evasion
- Viruses often reduce CpG content or use other strategies to avoid being flagged and degraded.
- Similar suppression mechanisms operate against incoming nucleic acids (e.g., in endosomes) and endogenous foreign‐like sequences, reinforcing the idea that GC/AT balance and the presence of introns serve as universal “fingerprints” of native versus unwanted RNA.
What can it not explain?
- CpG Islands vs. CpG Suppression
- Although mammalian cells often silence CpG-rich regions (for instance, via methylation or binding proteins such as ZAP), they also maintain CpG islands—regions dense in CpG dinucleotides that remain unmethylated and help activate transcription.
- Why the cell doesn’t simply methylate all CpG sites, and how it balances both promoting and suppressing CpG-rich regions, remains somewhat paradoxical.
- Exceptions Like L1 Transposable Elements
- Most successful transposable elements are GC-rich, which fits the hypothesis. However, L1 elements are GC-poor but still manage to evade numerous quality control filters.
- Part of the explanation is that L1’s ORF1 is shorter than the length threshold targeted by HUSH, but a deeper understanding of why L1 still transposes despite multiple suppression mechanisms is incomplete.
- No Quality Control Filter Is Perfect
- Some features—like intronless transcripts or those with high A content—are flagged as suspicious, yet there are essential genes (like histones) or transcripts in early embryogenesis that bypass or temporarily evade these checks.
- The hypothesis acknowledges that perfect suppression would eliminate even some beneficial transcripts, so “leaky” expression is unavoidable.
- Context-Specific Selection on Synonymous Sites
- Not all selection on synonymous codons revolves around blocking unwanted transcripts. Some mutations, for example, influence RNA structures like G-quadruplexes, or regulate binding by miRNAs. These do not neatly fit under the “unwanted transcript” explanation.
Tags: Codon usage, CpG, GC content
Categories: Paper digest
Comments